Faculty Research
Below is a sampling of Ray and Stephanie Lane Computational Biology Department research projects. Additional research projects are described on individual faculty homepages.
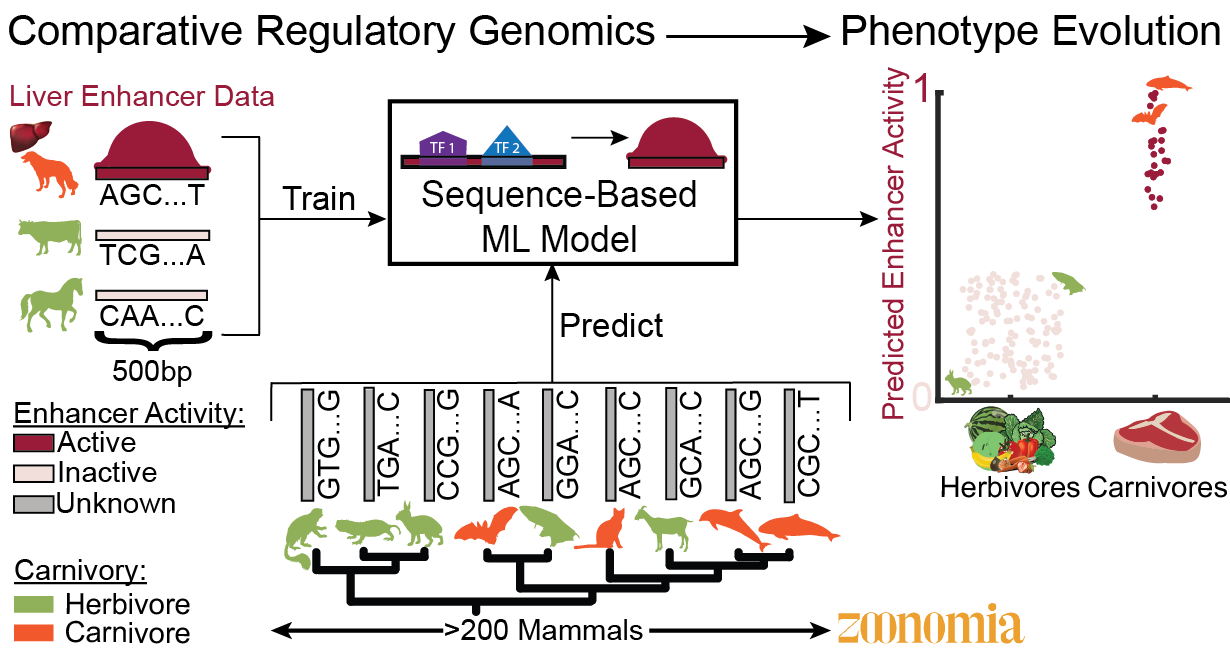 The Kaplow Lab studies vertebrate phenotype evolution by developing approaches for identifying enhancers associated with phenotype evolution that use machine learning predictions of tissue- or cell-type-specific enhancer activity across genomes from hundreds of species paired with species’ phenotype annotations. The results from our computational methods often lead to specific hypotheses about the roles of enhancers in regulating genes involved in phenotypes, and we test these hypotheses experimentally. We are currently focusing on metabolic phenotypes, such as carnivory and hibernation. We also investigate the specific mechanisms through which enhancer activity has changed between species, including the influence of changes in transcription factor binding that affect tissue-specific gene expression.
The Kaplow Lab studies vertebrate phenotype evolution by developing approaches for identifying enhancers associated with phenotype evolution that use machine learning predictions of tissue- or cell-type-specific enhancer activity across genomes from hundreds of species paired with species’ phenotype annotations. The results from our computational methods often lead to specific hypotheses about the roles of enhancers in regulating genes involved in phenotypes, and we test these hypotheses experimentally. We are currently focusing on metabolic phenotypes, such as carnivory and hibernation. We also investigate the specific mechanisms through which enhancer activity has changed between species, including the influence of changes in transcription factor binding that affect tissue-specific gene expression.
AI/ML for Spatial Biology in Health and Disease

The Ma lab has made several key recent breakthroughs that advance the frontier of computational biology in genome and cellular organization. They pioneered Higashi, Fast-Higashi, and scGHOST, a series of graph-based machine learning frameworks for analyzing single-cell 3D genome architecture (Nat Biotechnol 2022; Cell Syst 2022; Nat Methods 2024; Nat Genet 2024). In parallel, the lab introduced the concept of spatially variable metagenes to characterize tissue-level determinants of cell identity (Nat Genet 2023, Cover Article). Together, this body of work lays the methodological foundation for a next-generation Multiscale Cellular Model, with the potential to transform our understanding of complex biological systems in health and disease.
Active learning of cell organization
A team led by Bob Murphy, Ray and Stephanie Lane Professor of Computational Biology Emeritus and a faculty member in the Machine Learning Department, is combining image-derived modeling methods with active learning to build a continuously updating, comprehensive model of protein localization. The organization of eukaryotic cells into distinct organelles and structures is critical to cell health and function. Alterations in protein localization have been associated with many diseases, including cancer. Obtaining a complete picture of the localization of all proteins in cells and how it changes under various conditions is therefore an important but daunting task, given that there are on the order of a hundred cell types in a human body, tens of thousands of proteins expressed in each cell types, and over a million conditions (which include presence of potential drugs or disease-causing mutations). Automated microscopy can help by allowing large numbers of images to be acquired rapidly, but even with automation it will not be possible to directly determine the localization of all proteins in all cell types under all conditions. As described by Dr. Murphy in a recent Commentary in Nature Chemical Biology, the alternative is to construct a predictive model of protein localization from all existing information, and use it iteratively to identify those experiments that will are expected to maximally improve the model. This is an example of Active Learning, a powerful machine learning technique. The fundamental assumption when applied to biological experiments is that there are correlations between the results of experiments for different variables and that learning these correlations can allow accurate prediction of results for experiments that have not been (and may never be!) done.
Preliminary results in test systems have been very encouraging. Using active learning on data from Pubchem, a repository of results from high throughput drug screens, nearly 60% of drug “hits” were found while doing only 3% of all possible experiments. Encouraging results have also been obtained in extensive computer simulations of learning the effects of many drugs on many proteins. Based on these results, active learning-driven automated imaging of cell lines expressing different fluorescently-tagged proteins is currently being carried out in Dr. Murphy’s laboratory. The project draws on a range of cutting edge methods, and collaborators and consultants include a number of faculty members from the Machine Learning and Ray and Stephanie Lane Computational Biology Departments, including Chris Langmead, Aarti Singh, Gustavo Rohde, Jaime Carbonell, and Jeff Schneider.
Robert F. Murphy Group Software
-
-
- CellOrganizer – Open source system for learning and using generative models of cells from images.
- OMERO.searcher – Open source addon to OMERO to enable content-based image searching.
- PatternUnmixer – Open-source Matlab program for learning mixture models of subcellular patterns.
- SLIF – Structured Literature Image Finder – open source tools for extracting, analyzing and searching images from primary biomedical literature.
-
How The Genome Evolved for Vocal Learning and Speech Production
The Neurogenomics laboratory led by Assistant Prof. Andreas Pfenning is studying how the genome evolved for vocal learning, an aspect of speech production that sets us apart from other primates.
Although nothing is more central to our identity as human beings than behavior, we still don’t know how the billions of nucleotides in our genome have evolved to give us complex thoughts or the ability to communicate those thoughts through speech. The component of speech that gives us a window into the genetic basis of behavior is vocal learning, which is the ability to reproduce complex vocalizations. Although speech itself is uniquely human, vocal learning is an example of convergent evolution, having evolved independently in multiple species, including songbirds/parrots, hummingbirds, bats, elephants, and also in humans relative to chimpanzees. This allows the Neurogenomics Laboratory to take a comparative genomic approach to understanding how vocal learning evolved: what features do the genes and the genomes of vocal learning species have in common relative to those without the ability?
We approach this problem using a wide set of techniques. The Neurogenomics laboratory develops and adapts computational methods to identify regulatory elements in the genome associated with vocal learning behavior. We then use genomic techniques, such as high-throughput reporter assays and genome editing, to validate and refine the computational models. The results of these experiments will not only yield insights into how human speech evolved, but also provide a foundation for others who are aiming to connect genome sequence to organism traits using the wealth of new genomic data available.
The Immune Basis of Alzheimer’s Disease
The Neurogenomics laboratory led by Assistant Prof. Andreas Pfenning is studying how the immune system influences Alzheimer’s disease predisposition.
Alzheimer’s disease is a complex neurodegenerative disorder that has an enourmous impact on society, but is still poorly understood. Dr. Pfenning’s previous research has shown that sequence mutations associated with Alzheimer’s Disease are more likely to be found in regions of the genome that are active in the immune cells, and not within the brain. The goal is this research is to uncover how the immune system influences Alzheimer’s disease predisposition and progression.
We are tackling this problem by building computational models that combine human clinical data, large-scale genetics, epigenomics, and mouse models of neurodegeneration. These experiments aim to uncover biomarkers for Alzheimer’s disease and provide the foundation for immune-based therapeutics.
Ray and Stephanie Lane Computational Biology Department Head Russell Schwartz is directing a research project to apply methods from phylogenetics (i.e., the inference of evolutionary trees) to identify patterns of evolution of cell lineages within cancers. Cancer is a class of diseases defined by the accumulation of successive mutations that disable normal controls on cell growth, allowing rapid cell proliferation that typically results in tumor growth and eventually metastasis. A single tumor is therefore a kind of evolving population, characterized which rapid mutation and strong selection for cells that can proliferate quickly and evade normal cellular controls on growth or anti-tumor therapies. This observation led to an approach called tumor phylogenetics (Desper et al., 1999) in which one applies phylogenetic algorithms to genetic profiles of tumors to infer evolutionary trees that describe the common features of progression across tumors. These trees then describe the sequences of mutations, also known as progression pathways, that characterize common tumor types. These tumor phylogenies can help researchers identify common features among seemingly different tumors, discover particularly important steps in evolution, and gain insight into the mechanistic reasons why particular patterns of progression seem to recur across many patients. It is hoped that such insights will help us identify new targets for anti-tumor drugs and develop better diagnostic tests to determine which patients are likely to benefit from which therapies.
This work arose from a collaboration between Schwartz and Dr. Stanley Shackney and is now being carried out in collaboration with Alejandro Schäffer, Kerstin Heselmeyer-Haddad, and Thomas Ried of the National Institutes of Health. A particular focus of their work is the question of cell-to-cell heterogeneity in single tumors. The Schwartz lab has pursued this problem from several directions. These include the use of direct single cell data, through a special focus on the use of fluorescence in situ hybridization (FISH) data as well as algorithmic work on the use of single-cell sequence data sets. A second direction is the use of deconvolution (unmixing) methods to reconstruct the genomics of cell subpopulations from bulk genomic data. There work spans modeling and algorithmic directions in better representing the process of tumor evolution and heterogeneity and improving algorithms to fit data to those models, machine learning methods for applying these reconstructions to predicting tumor progression and patient prognosis, and application of these methods with research collaborators.
A talk by Schwartz at the Simons Institute for Theoretical Computer Science on some of his directions in FISH-based tumor phylogenetics is shown below.